

医薬品開発のプロセスを解説!期間・費用・今後の展望も紹介

医薬品開発事業に新規参入する場合、事前に医薬品開発のプロセスや関連法令、課題を把握する必要があります。
本記事では、医薬品開発のプロセスや開発に必要な期間、費用、関連法令、現状の課題と展望を解説します。医薬品開発ベンチャーの立ち上げを検討中の方や、他業界・他業種の企業を経営していて医薬品開発分野に進出しようとお考えの方は、ぜひ最後までご一読ください。
医薬品開発のプロセス
医薬品開発は、大まかに以下の流れで進みます。
- 基礎研究
- 非臨床試験
- 臨床試験
- 申請・審査・承認
- 販売
- 調査
以下で、各プロセスに関して詳しく説明します。
①基礎研究
基礎研究は、医薬品の候補に適した物質を探したり合成したりする段階です。天然化合物や人工的に合成された化合物に関して、主に動物由来の細胞などを用いて活性を調べ、候補物質を幅広く探索します。
活性がある物質(リード化合物)が発見されたら、さらに活性が強い化合物を求めてリード化合物に修飾を施し、多様な誘導体が作られます。
なお、基礎研究の段階では、主に薬効に焦点を当てて研究が実施されます。
②非臨床試験
非臨床試験(前臨床試験)は、基礎研究で絞り込まれた化合物について、動物や培養細胞などを用いて有効性や安全性を評価する段階です。薬理作用・毒性・薬物動態などを確認し、人に投与した際の有効性や安全性を予測します。具体的には、安全性を評価するための毒性試験(一般毒性試験)や薬理試験(薬効薬理試験)に加え、体内での吸収・分布・代謝・排泄(ADME)を確認する薬物動態試験も行われます。各試験の結果をもとに、有効用量と毒性(特殊毒性試験、安全性薬理試験)が現れる用量の関係を見極め、臨床試験へ進めるかどうかを判断します。
多くの製薬会社では、この段階から、研究開発部門だけではなくマーケティング・渉外など、様々な部門のトップが出席する最高意思決定機関で方針を決定します。
③臨床試験
臨床試験(治験)は、有効性・安全性を確認するために、ヒトを対象に治験薬(開発候補品)を投与する段階です。臨床試験は、以下の3フェーズで構成されます。

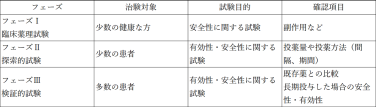
なお、臨床試験は、医薬品開発の全プロセスのなかで最も長い時間がかかるプロセスです。非臨床試験から承認まで、一般的に10~15年程度の期間を要します。
ただし最近は、コロナワクチンのように緊急使用のため特例承認という形で使用され、その使用実績が有効性・安全性を評価するデータとして扱われ、正式に承認されることもあります。しかし、これはあくまで特例であり、通常の医薬品開発には長期間を要します。また、莫大な資本が必要で、成功率も極めて低く、多くがフェーズIIの段階で開発中止になっています。
④申請・審査・承認
臨床試験が無事に終了したら、厚生労働省に対して医薬品の承認申請を行います。申請後、PMDAや、専門家によって成分・分量や用法・用量、効能・効果、副作用などに関する所要の審査が実施され、承認されると薬価が決定され、薬価基準に収載されます。
申請に必要な資料や申請方法の詳細は、厚生労働省の公式サイトなどから確認できます。事前に、独立行政法人医薬品医療機器総合機構(PMDA)に相談して対応策を協議、確認して申請作業を進めてください。申請資料の内容に関して不十分な点を指摘してもらえれば、資料提出の際に改善した資料を提示でき、手続きの効率化につながります。
⑤販売
薬事・食品衛生審議会の審議を経て厚生労働大臣に承認されると、医薬品として製造・販売が可能になります。
ただし、新医薬品は治験の症例数に限りがあり、市販後に多くの患者へ使用された段階で、治験中には把握しきれなかった副作用や使用上の課題が明らかになる場合があります。
そのため、新医薬品には、承認後の一定期間に実際の使用成績を収集し、有効性や安全性を改めて確認する再審査制度が設けられています。フェーズIV(第4相臨床試験)は、市販後臨床試験として位置付けられます。
⑥調査
販売開始後も、継続的に安全性(副作用・使い方など)に関する市場調査を実施する義務があります。何らかの問題点が判明した場合は、直ちに社内で協議し、重大な場合はPMDAへ届け出た上で解決策を協議し、改良の検討を開始します。
問題点の解決に至った場合は、報告書を作成してPMDAに報告し、承認を得たのち、社内手続きで変更管理を行います。その上でQAの承認を得て、改良法で得られた製品の品質同等性を確認することにより、その後、次のステップへ進み、製造・出荷が可能となります。
より安全で使いやすい医薬品に改善した場合は、新規化合物として扱われ、試験をやり直す必要が生じる場合があります。改良したからといって簡単に製造・販売できるわけではありません。この点は、医薬品を取り扱う上で特に注意が必要です。
多くの製薬企業では、医薬情報担当者(MR、Medical Representative)が、安全性に関する情報収集業務を担当します。
医薬品開発に必要な期間
医薬品開発に必要な期間は、全段階の合計で9~17年程度です。以下では、開発段階ごとに必要とされる期間の目安を示します。
- 基礎研究:2~3年
- 非臨床試験:3~5年
- 臨床試験:3~7年
- 申請・審査:1~2年
また、開発成功率の推移は以下のとおりです。
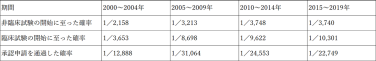
年々、医薬品開発の成功率は低下し、難易度は上昇する傾向にあります。
医薬品開発にかかる費用
医薬品開発には、数百億円から数千億円規模の費用がかかるとされています。以下では、医薬品開発の費用が高額になりやすい理由と、費用負担を抑えるための取り組みを解説します。
医薬品開発の費用が高額になる理由
医薬品開発の費用が高額になりやすい主な理由は、開発に長い年月を要する上、多くの候補化合物が途中で開発中止となるためです。
医薬品の開発では、基礎研究から非臨床試験、臨床試験、承認申請まで複数の段階を経る必要があり、各段階で試験や評価を重ねるため、研究開発費が積み上がります。
最終的な開発コストには、開発途中で中止となった候補化合物にかかった費用が含まれることも、医薬品開発が高額になる理由のひとつです。
また、近年はバイオテクノロジーの進展により、低分子医薬品に加えて抗体医薬など高分子医薬品や核酸医薬のような中分子医薬品と開発対象の幅が広がり、医薬品のプラットフォーム選択や技術開発の重要性が高まっています。
このような変化を受け、医薬品開発は従来よりも複雑化しており、必要となる知見や技術、投資の面でも負担が大きくなる傾向にあります。
医薬品開発の費用を抑える取り組み
医薬品開発では、コスト増大に対応するため、開発プロセスの効率化につながる取り組みが進められています。
代表的な取り組みのひとつが、AIの活用です。AIを活用して大量の生体内分子データやオミクスデータを解析できれば、疾患に関わる分子や創薬標的の特定、候補化合物の探索・設計、薬物動態や毒性予測の効率化に役立ちます。データ収集・解析へのAIの活用は、今後さらに広がることが予想されます。
こうした技術の活用は、実験前の選別精度を高め、創薬にかかる時間とコストの削減につながります。
また、外部の企業や研究機関との連携も有効な選択肢です。近年は、研究機関や創薬ベンチャー、製薬企業、CDMOなどが役割を分担する「水平分業型」の創薬が進み、コストの削減や開発の迅速化、専門人材の活用による成功確率の向上などが期待されています。
医薬品開発の費用負担を抑えるためには、自社で全てを抱え込むのではなく、データ活用によって研究開発の精度を高めつつ、外部連携を通じて必要な機能を補完する視点が重要です。
医薬品開発に関する法令・ルール

医薬品の研究開発では、被験者に健康被害が生じる可能性があります。また、販売後に欠陥が判明し、患者に健康被害が発生すると大きな社会的影響が生じるため、数多くの倫理規範・ガイドラインなどが定められています。
これまで、ニュルンベルク綱領(1947年)や世界医師会によるヘルシンキ宣言(1964年)、米国ベルモント報告(1979年)、欧州臨床試験指令(2001年)、国際医学団体協議会(CIOMS)によるガイドライン(2002年)など、数多くの規範・ガイドラインが策定され、臨床研究に関する原則が形作られました。
現在の日本では、以下に示す法令などにより、治験の規制が実施されています。
- 医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(薬機法)
- 医薬品の臨床試験の実施の基準に関する省令(GCP)
- 医薬品の安全性に関する非臨床試験の実施の基準に関する省令(GLP)
- 医薬品及び治験薬の製造管理及び品質管理の基準(GMP/治験薬GMP)
- ICH Q7原薬GMPガイドライン第19章:臨床試験に使用する原薬
これらの法令に違反すると、刑事罰が科されるケースもあるので注意が必要です。例えば、承認前医薬品の広告・宣伝を行うと、2年以下の拘禁刑もしくは200万円以下の罰金が科される(または、併科される)可能性があります。
法令に違反すると承認が得られず、せっかく開発した医薬品も販売できません。事前に各種法令を熟読し、不明な点がある場合は厚生労働省やPMDA、専門家などに相談しましょう。
日本国内における医薬品開発の現状と展望
以下、日本国内における医薬品開発の現状と課題、および、今後の展望に関して説明します。
医薬品開発の現状と課題
1993年時点では、日本の大手製薬会社の研究開発費は、アメリカの大手製薬会社の研究開発費の36%程度でした。2019年時点では、約22%の水準にまで下落しています(※)。
以下は、世界の製薬企業の売上高と開発費(2020)の例です。世界の製薬企業と日本の製薬企業の規模を比較できます。

また、近年、世界各国でベンチャー企業による医薬品開発が活発ですが、日本では創薬ベンチャーによる開発事例は多くありません。その理由のひとつは、日本の創薬ベンチャーに集まる資金が少ないことです。ベンチャーから短期間で規模を拡大した例は、特にアメリカに多く見られます。
例えば、コロナワクチンを開発したモデルナ社や、現在では大手製薬企業として存在感を示すギリアド・サイエンシズ社も、元はベンチャー企業でした。優れた製品を開発できる企業は、急成長できることを示しています。
※出典:厚生労働省「医薬品産業の現状」
今後の展望
近年、世界各国で創薬ベンチャーが活躍していますが、日本ではベンチャー企業による医薬品開発が低調です。この実情を受け、日本政府はベンチャー企業による開発を支援するために多種多様な施策を実施しています。以下は、施策・取り組みの具体例です。
- 産学官共同創薬研究プロジェクト(GAPFREE)
- 全ゲノム解析等実行計画
- バイオコミュニティ形成支援
- 資金提供
- 税制優遇
産学官共同創薬研究プロジェクトは、アカデミア(大学・研究機関)が保有する知見・技術と、製薬会社の創薬ノウハウをつなげる取り組みです。また、全ゲノム解析等実行計画は、難病の効果的な治療法の開発に役立ちます。
産業界・大学・自治体が参画するバイオコミュニティ形成支援も重要な施策のひとつです。人材・投資の呼び水となる支援策を継続的に追加・充実させる方針が示されているため、今後、日本発の医薬品の増加が期待されています。
今後は、大学教育の抜本的改革やサイエンティスト育成プログラムの充実、継続的な資金援助が重要になるでしょう。日本発のベンチャーが有望な製品を開発した場合、海外企業によるM&Aの対象となる可能性もあります。国による継続的な経済支援が期待されます。
なお、バイオ医薬品のCMO/CDMO市場は成長を続けており、年率8%程度の成長が見込まれています。現時点では、グローバル市場でスイス・韓国・ドイツの企業が活躍しており、日本企業の存在感は大きくありません。しかし、日本国内では、新たな会社の設立やM&Aなどによる既存事業の拡大や新規事業の立ち上げが進み、ここ数年、積極的な投資が急速に進んでいます。
CDMOに関してより詳しく知りたい方は、以下の記事もあわせてご覧ください。
▶関連記事:CDMO(医薬品開発製造受託機関)とは?CMOとの違いや活用するメリットを解説
医薬品開発に関連する情報やサービスを探すなら「ファーマラボEXPO」へ
RX Japanが主催する「ファーマラボEXPO」は、インターフェックスWeekの構成展のひとつで、医薬品研究開発向けの支援サービスや最新機器が展示される展示会です。医薬品開発に従事する方は、ファーマラボEXPOにご来場の上、最新情報を収集してはいかがでしょうか。
また、ファーマラボEXPOは、支援サービスや機器を開発・販売する企業にとっても、自社の技術やサービスをPRする有益な展示会です。取引先の新規開拓をしたい方は、ブースを出展してみてはいかがでしょうか。
なお、入場には来場登録が必要です。登録方法については、以下のページをご確認ください。




プロセスの流れや課題を把握して、医薬品開発に取り組もう
医薬品の開発プロセスは、基礎研究、非臨床試験、臨床試験(治験)の順番で進行します。その後、厚生労働省に承認申請を実施し、審査に通過したら医薬品として製造・販売が可能です。なお、副作用などに関する調査は、販売開始後も継続的に実施する必要があります。
医薬品の開発には長い年月がかかり、成功率は決して高くありません。また、各種法令を遵守する必要もあります。
近年は、AIを活用した研究開発の効率化や、外部企業・研究機関との連携による役割分担など、費用負担を抑えるための取り組みも重要になっています。日本政府は、創薬ベンチャーを育成するための施策を実施しているので、今後の動向に注目したいところです。
ファーマラボEXPOでは、医薬品開発向けの支援サービスや最新機器が展示されます。医薬品開発に携わっている方はファーマラボEXPOにご来場の上、最新の情報を収集してはいかがでしょうか。支援サービスや機器を開発・販売する企業にとっても、ファーマラボEXPOは有益な展示会です。販売先の新規開拓につながるので、ぜひ出展をご検討ください。

西日本最大級!業界のトレンドがわかる3日間


医薬品・化粧品・再生医療に関する専門技術展
会期:2026年9月30日(水)~10月2日(金) 会場:インテックス大阪

▶監修:橋本 光紀
医薬研究開発コンサルテイング 代表取締役。
九州大学薬学部修士課程修了後、三共株式会社の生産技術所に入社し研究に従事。その後、東京工業大学で理学博士号を取得し、M.I.T.Prof.Hecht研・U.C.I.Prof.Overman研に海外留学。
1992年よりSankyo Pharma GmbH(ドイツ、ミュンヘン)研究開発担当責任者となり、2002年には三共化成工業(株)研究開発担当常務取締役となる。
2006年に医薬研究開発コンサルテイングを設立し、創薬パートナーズを立ち上げ現在に至る。
▼この記事をSNSでシェアする